ARJ 2017, 1(1), P.P. 1-22;
archives/2017/Ahmed_Alshather/Cancer_and_chemotherapy/
Review Article
Cancer and chemotherapy review
Ahmed Ismail Alshather; Ph. D. Pharm. Chem, College of Pharmacy, University of Baghdad*
Introduction
Cancer is a class of diseases characterized by uncontrolled division of cells and the ability of these cells to invade other tissues, either by direct growth into adjacent tissue (invasion) or by implantation into distant sites (metastasis). This unregulated growth is caused by damage to DNA, resulting in mutations to genes that control cell division. Several mutations may be required to transform a normal cell into a malignant cell. These mutations are often caused by chemicals or physical agents called carcinogens. Some mutations occur spontaneously, or they can be inherited (germ line mutations)1.
Using chemicals to treat cancer, it has played a major role in cancer treatment for half a century. Years of testing and research have provided chemotherapy to be an effective cancer treatment. It may be the only treatment, or it may be used in combination with other treatments, such as surgery and radiation therapy2. Chemotherapy works by killing rapidly divided cells. These cells include cancer cells, which continuously divide to form more cells, and healthy cells that divide quickly, such as those in bone marrow, gastrointestinal tract, reproductive system and hair follicles. So, chemotherapy does not know the difference between the cancerous cells and the normal cells. The normal cells will grow back and be healthy but in the meantime, side effects occur3.
↑|ahmedalshather|@|yahoo|.|com
Types of Chemotherapy
Since the discovery of mustard gas, as the first anticancer agent4, many anticancer compounds with different modes of action have been discovered and studied. These agents can be roughly classified as follows5:
- Alkylating agents. These medications interfere with the growth of cancer cells by blocking the replication of DNA. They contain chemical group that can form covalent bond with particular nucleophilic substance in the cell6. Most of cytotoxic anticancer alkylating agent are bifunctional (i.e., they have two alkylating groups)7. Some of the more common alkylating agents include Cyclophosphamide, Melphalan, Chlorambucil, Decarbazine, Procarbazine, Busulfan, and Thiotepa.
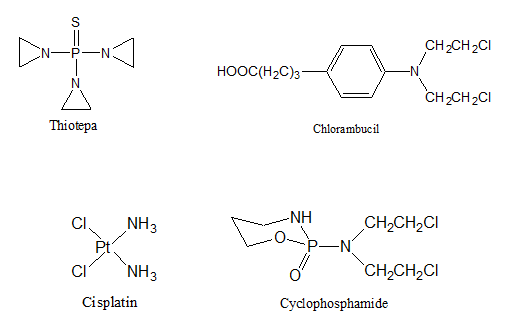
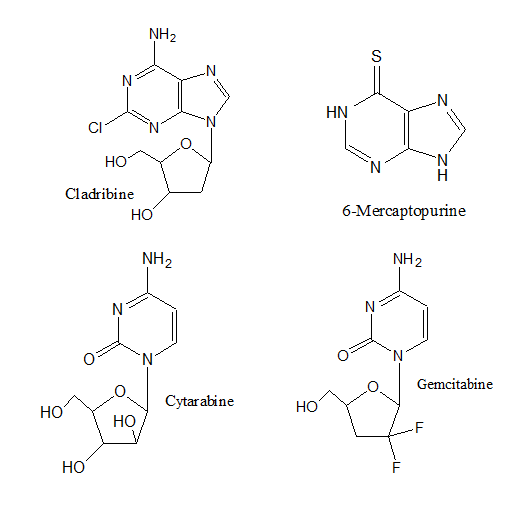
- Antimetabolites. These drugs block the enzymes needed by cancer cells to live and grow. They are usually closely related to the metabolite that is antagonized. These agents may have the capacity to combine with site of action as if they are substrates or cofactor8. Alternatively, they may bind to an allosteric regulatory site, especially when they resemble the end product of biosynthetic pathway under feed back control9. Some antimetabolites that inhibit DNA synthesis and DNA repair include: Cytarabine, Gemcitabine (Gemzar®), 6-mercaptopurine, 6-thioguanine, Fludarabine, and Cladribine.
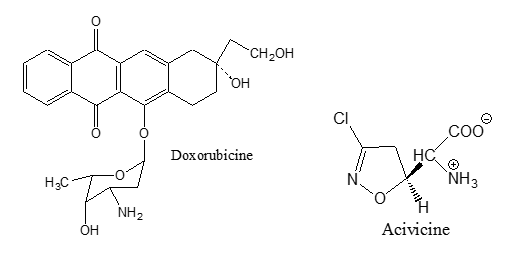
- Anti-tumor antibiotics. These are a number of compounds which are first isolated from bacterial cultures to be used as antibacterial agents, but these compounds were originally rejected as antibacterial agents because of their cytoyoxicity10. Later, it was found that this toxicity could be turned to an advantage in chemotherapy of cancer11. They interfere with DNA, blocking certain enzymes and cell division and changing cell membranes12. Doxorubicine13 Mitomycine14 and Acivicin15 are examples of these antibiotics.
Other types of anticancer agent include plant derivative (Vincristin, vinblastine)16, hormones (Tamoxifen17, Flutamide18) and nitrosoureas (Carmustine19, Lamustine20). Chemotherapy medications, regardless of how they are given, generally travel in the bloodstream and throughout the entire body. The intravenous route is the most common, allowing chemotherapy drugs to spread quickly through the system.
Chemotherapy goals
One of chemotherapy's main advantages is that — unlike radiation, which treats only the area of the body exposed to the radiation — chemotherapy treats the entire body. As a result, any cells that may have escaped from the original cancer are treated21.
Chemotherapy used to:
- Eliminate all cancer cells in the body, even when cancer is widespread.
- Prolong the life by controlling cancer growth and spread.
- Relieve symptoms and enhance the quality of life.
Side effects of chemotherapy
Chemotherapy does not only target cancer cells, but any cells that divide rapidly. This includes those in the bone marrow and skin, hair-producing cells, and the cells that line the mouth and digestive system. Damage to these healthy cells may produce various temporary side effects, including22:
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
How long these temporary side effects last depends on what drug(s) were taking and for how long. Most side effects will subside shortly after stopping chemotherapy treatments. In addition, most short-term side effects can be minimized with medication. As people with cancer are living longer after treatment, doctors have discovered that some treatments cause long-lasting side effects or side effects that become apparent long after treatment ends. These long-term side effects are 23:
- Organ damage, including problems with the heart, lungs and kidneys
- Nerve damage
- Blood in the urine (hemorrhagic cystitis)
- Another cancer, including Hodgkin's disease and non-Hodgkin's lymphoma, leukemia and some tumors
Improvement of current chemotherapies
Selective delivery of lethal concentrations of drugs to tumors, allowing the latter to be eradicated without damage to other tissues, continues to be a major goal in cancer chemotherapy24. In order to improve the efficacy of current chemotherapeutic treatment and diminish side effects, prodrug strategies have evolved in an attempt to achieve site-specific delivery of cytotoxic anticancer agents. This strategy is based on the relative lack of toxicity of prodrugs in comparison to their anticancer (chemotherapeutic) drug counterparts25. Such strategies often take advantage of differences between tumor cells and "normal" healthy cells, based on tumor-associated factors, such as hypoxia, tumor-associated enzymes and receptors. Compartmental release of the drug inside cells can be achieved by designing the spacer so that it is stable outside the cells, but quickly degraded in the appropriate compartment. Two possible parameters to exploit include the difference in pH26 and presence of enzymes in endosomes and lysosomes27,28. Any of a number of non-toxic prodrugs may be converted to a toxic chemotherapeutic agent by the action of an enzyme. Exemplary enzymes that may be used to convert a prodrug into the form of an active chemotherapeutic agent include, but are not limited to, cytosine deaminase, nitroreductase, β-glucuronidase, β-galactosidase, a carboxypeptidase, alkaline phosphatase and β-lactamase29. In addition, at least two carboxypeptidases have been used to convert a prodrug into a chemotherapeutic agent, e.g. the conversion of methotrexate-α-alanine to methotrexate30. Carboxypeptidase G2 used in a gene-directed enzyme prodrug therapy (GDEPT) system, for use in the conversion of nitrogen mustard prodrugs into the corresponding active form31. Carboxypeptidase G2 finds utility in the cleavage of amidic, urethanic or ureidic bonds32.
Polymeric drug delivery
Although increasing numbers of new pharmaceuticals are macromolecules, the vast majority of the current therapeutics consists of small molecular weight compounds. Several advantages often lead to small molecules being chosen over macromolecules for new therapeutics. Combinatorial chemistry is well suited to produce a large number of derivatives of small molecules, reducing the time and cost to find an active agent33, 34. Once an active compound is discovered, a compromise between activity, solubility, and permeability must be found. Often, highly active agents are abandoned because simple chemical or formulation modifications cannot produce a compound with sufficient solubility and permeability while maintaining high activity. These and other potential problems can often be overcome by conjugating drugs to water-soluble polymers. Advantages of polymeric drug delivery include increased solubility of hydrophobic drugs; passive and active targeting; altered protein binding, biodistribution, pharmacokinetics, and pharmacodynamics; as well as controlled release of drugs35. A suitable polymeric carrier can turn an unstable, insoluble, immunogenic drug with many toxic side effects into a highly specific and effective therapeutic agent.
Polymeric controlled drug delivery systems
There are two main types of polymer drug delivery systems; polymeric controlled release systems and polymer-drug conjugates. Since the impetus of this thesis is polymer conjugates, polymeric controlled release systems will only be briefly reviewed. Polymers can be used to provide controlled release of drugs36,37 . The ideal controlled release system would maintain drug levels in a therapeutically desirable range—whether that is at a constant level, rhythmic circadian levels, or responsive to specific conditions. This could greatly reduce side effects, effectively delivery drugs with short half- lives, potentially decrease the amount of drug needed, decrease the number of dosages, and increase patient compliance. This could not only save money, it could save lives. It was estimated that up to 15% of all hospital admissions, 136 billion $ in health care costs, and 100,000 deaths in the US each year can be linked to adverse drug events. An additional 10% of hospital admissions are attributed to a lack of patient compliance. Side effects could also be decreased due to localized release of the drug. Several types of drug delivery strategies have been conceived to achieve controlled release of drugs such as thermo, chemical, and pH sensitive hydrogels; biodegradable polymer; drug coatings; liposomes; and osmotic pumps38.
Polymer drug conjugates
Drugs can also be bound to polymers yielding polymer-drug conjugates. Conjugation of drugs to water-soluble polymers can overcome many different types of problems, including increasing the solubility, lowering the toxicity, improving the pharmacokinetics and pharmacodynamics through prolonged circulation times and an altered biodistribution, increasing the stability of the drug, decrease immunogenicity, and active and passive targeting of the conjugate39.
Drugs conjugated to macromolecules behave differently from the parent drug. Care must be taken in choosing the polymer with the correct properties to avoid interactions that may decrease efficacy as well as introduce other incompatibilities. An example would be the conjugation of too many hydrophobic drugs resulting in intermolecular or intramolecular aggregation leading to altered solubility, biorecognition, and biodistribution40,41. These can potentially result in a decrease in the therapeutic efficacy. Care must be taken when choosing the polymer to avoid intrinsic effects as well as maximize delivery to the active site. Properties that should be considered include biocompatibility, toxicity, immunogenicity, solubility, biodegradability of the polymer, and ability of the drug to be released from the polymer42.
One of the most significant changes for a drug conjugated to a polymer is the biodistribution. For low molecular weight compounds, which are the majority of the current pharmaceuticals, the biodistribution is primarily controlled by diffusion, surface charge, and hydrophobicity. Polymers are typically much larger than the compounds they are attached to and mask out the properties of the small drug. Even when the drug is a macromolecule, such as a large protein, polymers conjugated to the drug will coat the surface and mask the drug's properties provided the density is sufficiently high. Thus, the properties of the polymer will most often dominate the polymer conjugate's properties resulting in changes in the biodistribution, pharmacokinetics, pharmacodynamics, etc.43. The diffusion of macromolecules through biological membranes is severely limited and these compounds can typically enter cells only by endocytosis44. If the polymers are larger than the renal threshold (a hydrodynamic radius of approximately 45 A°)45, elimination via glomerular filtration is reduced or halted resulting in an increased blood half-life.
Many drugs have low water solubility. Conjugation to water soluble polymers increases their solubility—which can often be reason enough to use polymeric delivery. Polymer conjugation can also alter the amount of protein binding and other affinities. This can not only affect the blood half-life, but also lead to large changes in the tissue distribution. This, in turn, can change the severity, number, and types of side effects as well as the therapeutic effect35.
Drug targeting
Most drugs currently in use typically have a wide distribution throughout the body. To achieve a therapeutic concentration at the target compartment, large doses must be given with the majority of the drug residing in normal tissue. The drug molecules interact with many organs and cells often triggering undesirable side effects. Targeted drug delivery involves the selective accumulation of a compound in a pathological organ, tissue, or cell where it will have the greatest effect. This concept of a guided missile or a "magic bullet" in drug delivery was described by Ehrlich in 190646. In 1975, Ringsdorf first described a comprehensive rationale for the use of polymers as targetable drug carriers47.
Targeting a drug or a drug delivery system results in a large change in the biodistribution—regardless of whether the targeting is active or passive. For toxic drugs, a reduction in side effects is often the greatest advantage of targeted delivery. The reduction in side effects is due to the targeted drug having a much higher chance of interacting with only its target coupled with a lower possibility of residing and interacting in nontargeted organs or compartments. Since a larger fraction of the administered dose reaches its target, lower doses can be given further reducing the possibility of side effects as well as reducing the cost of the expensive therapies35,38.
The various methods for targeting polymers will be briefly reviewed and can be grouped into four main ways:
- Direct application of the drug to the affected site48.
- Physical targeting based on properties such as abnormal pH or temperature49,50 or magnetic targeting51,52 via paramagnetic drug carriers,
- Passive accumulation.
- Active targeting using ligands or other vectors.
In this research Passive accumulation will be considered. Passive targeting involves changing the distribution of a compound without the use of a specific targeting moiety. The altered distribution is due to a change in the size, charge and hydrophobicity from that of the parent compound, the Enhanced Permeability and Retention (EPR) effect is an example of passive targeting (Fig.1). For polymers with a molecular weight >100,000, the extravasation of the polymer into normal tissue is limited. In contrast, tumors have blood vessels with increased permeability53,54 as well as a defective vascuiature55,56 that allows macromolecules to permeate into tumors. The size of material, which can penetrate this "leaky" vasculature, can vary and may depend on several other properties such as the charge and hydrophobicity / hydrophilicity57. Extensive angiogenesis also results in increased tumor vascuiature density58,59.
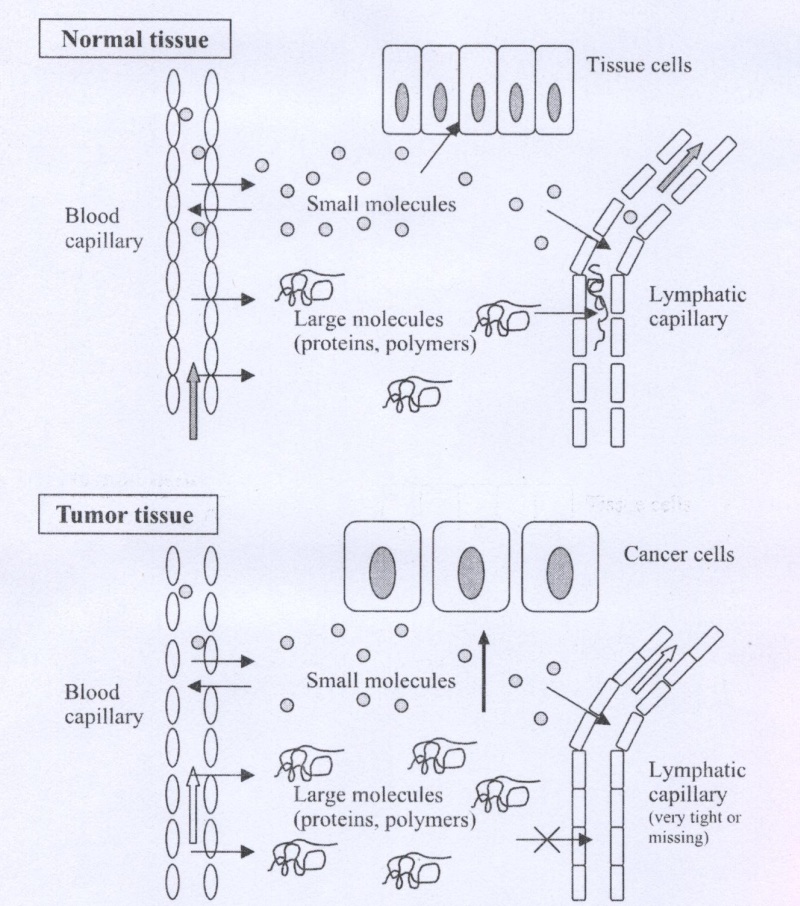
These factors combine to result in enhanced permeability of macromolecules in the tumor.
Fig.1. Schematic illustration of the EPR effect principle. Angiogenesis and enhanced vascular permeability of tumor capillaries and impaired or missing lymphatic clearance of macromolecules result in accumulation of macromolecules (polymers) in tumor tissue.
Inflamed tissue also has increased permeability, but macromolecules and lipids in the interstitial space are rapidly cleared via the lymphatic system. Tumors have a defective lymphatic drainage which results in very slow clearance of macromolecules and lipids from the tumor interstitium60. The slow clearance of macromolecules is termed "enhanced retention" and when combined with the enhanced permeability yield an increased distribution of macromolecules in tumors compared to other tissue.
Thus, by simply increasing the molecular weight of the anticancer drugs via polymer conjugation, passive targeting is achieved.
Other way to target a nonspecific drug is by binding of a ligand or other vector capable of specific recognition of the target site.
Numerous targeting, homing device (Fig. 2), moieties can be used including antibodies and antibody fragments, mono-, oligo-, and poly-saccharides, lectins, lipoproteins, hormones, other proteins, charged molecules, and some low molecular weight ligands such as folate49. The efficacy of the targeting system depends upon the binding constant and the distribution of the receptors throughout the body.
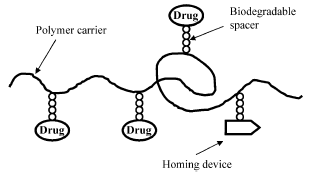
Fig. 2. Scheme of a polymer prodrug.
Care must be taken to assure that high drug loading does not result in aggregation or reduced activity. Conjugating the drugs to the polymers via biodegradable bonds allow release of the drugs .If the targeting moiety is an antibody or antibody fragment, binding it to a polymer often lowers its immunogenicity61,62.
Properties of polymeric carrier
In general, the polymer should be easy to synthesize and characterize. The polymer and its metabolites should be nontoxic and nonantigenic. Its molecular weight and structure should protect the drug from fast elimination processes (urinary excretion, hepatic uptake), it should contain functional groups permitting covalent binding of the drug and targeting moiety, it should be watersoluble, biocompatible, nonimmunogenic, the spacer (and possibly also the polymer) has to be biodegradable and the synthesis should be reproducible and inexpensive. Polymers suitable for preparation of polymer prodrugs can be categorized according to their origin (natural or synthetic), chemical nature (vinyl and acrylic polymers, poly ethylene glycol, polysaccharides, poly amino acids, etc.) and the backbone stability (stable or biodegradable). The biodegradable spacer has to be tailor-made either as a substrate for any lysosomal enzyme (lysosomotropic drugs) or susceptible to chemical hydrolysis63.
The water-soluble polymer-based drug carrier systems enable reduction of renal excretion of drugs and hence their better bioavailability, passive accumulation in solid tumors and, in the optimum case, controlling the locality and rate of drug release. A large variety of anti-cancer drugs have been covalently attached to many natural or synthetic water-soluble polymers using acid-sensitive linkage. Among natural polymers, polysaccharides such as dextran, inulin, pullulan or chitosan, proteins such as human and bovine serum albumin, transferrin or selected glycoproteins have been most frequently employed as drug carriers64. The advantage of natural macromolecules is their low toxicity, biodegradability and availability.
A certain drawback consists in their poor batch-to-batch reproducibility, in some cases immunogenicity and changes in their biodegradability and other properties after chemical modification. Synthetic polymers are a second group of drug carriers characterized by broad structure variability, good reproducibility of their synthesis and easy modification. In some cases, nondegradability could be a disadvantage of such carriers65.
Acid-sensitive polymer–drug conjugates
The hydrazone bond formed between the C13 carbonyl group of anthracyclines (Dox, daunomycin (Dau)) and polymer hydrazides66,67, or amide bond of a cis-aconityl residue containing spacer68 have been most often used for preparation of polymer–drug conjugates. Also there has been the use of spacers containing substituted trityl, acetal or imino groups69.
Shen and Ryser70 were one of the first who elaborated the concept of pH controlled drug release using modified polyacrylamide beads via cis-aconityl spacers. The greatest challenge of the acid-sensitive spacer was achieving relative stability at pH = 7.4, but quickly degrading at a pH between (5 – 6) to allowing a chance for hydrolysis in cancer tissue that have acidic environment (pH= 5.5-6.5)71.
Polymer-drug conjugates in clinical development
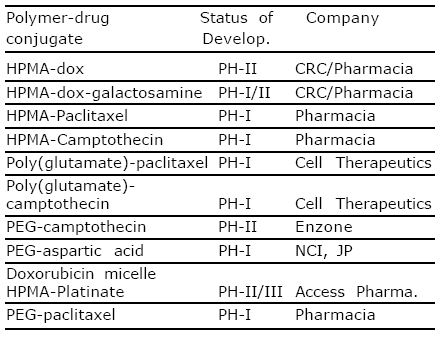
Conjugates that have entered clinical trials are listed in table (1)72.
Table 1. Polymer drug conjugate in early clinical trial as anticancer agents
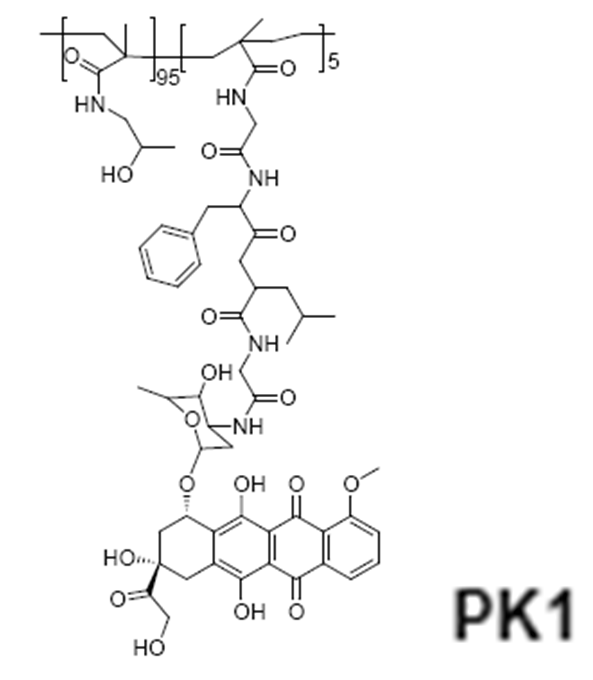
HPMA copolymer-doxorubicin (PK1, FCE28068)
The first synthetic polymer-drug conjugate to enter clinical evaluation was N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer-Gly-Phe-Leu-Gly-doxorubicin (PK1, FCE28068) in 199473. The homopolymer poly- HPMA was originally developed in the Czech Republic as a plasma expander8. It was nontoxic in preclinical tests at doses up to 30 g/kg, did not bind blood proteins, and was not immunogenic. Phase I/II clinical trials involving N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer-doxorubicin (PK1; FCE28068) showed a four- to fivefold reduction in anthracycline-related toxicity. Despite, cumulative doses up to 1680 mg/m2 (doxorubicin equivalent), no cardiotoxicity was observed. Antitumour activity in chemotherapy-resistant/refractory patients (including breast cancer) was also seen at doxorubicin doses of 80–320 mg/m2, consistent with tumour targeting by the enhanced permeability (EPR) effect.
HPMA copolymer-doxorubicin-galactosamine (PK2, FCE28069)
PK2, FCE28069 was the second compound emanating from CRC-funded research program. FCE28069 contains doxorubicin bound to the polymer backbone via a Gly-(D,L)Phe-Leu-Gly linker and it also contains galactosamine, included to promote liver targeting. Endocytic uptake by hepatocytes after interaction with the asialoglycoprotein receptor (ASGR) leads to lysosomal trafficking and their subsequent degradation. PK2 was designed with the aim of improving treatment of primary hepatocellular carcinoma and metastatic liver disease74.
PK2, FCE 2806
HPMA copolymer-paclitaxel (PNU166945)
The poor water solubility of paclitaxel combined with hypersensitivity reactions associated with the standard ethanol and cremophor formulation made it a good candidate for polymer conjugation. Pharmacia developed an HPMA copolymer conjugate of paclitaxel, with the aim of improving drug solubility and subsequent “controlled release” of paclitaxel. The resulting conjugate (PNU166945) was more soluble than paclitaxel (more than 2 mg/mL conjugate compared to 0.0001 mg/mL paclitaxel) and had a drug content of approximately 5% w/w. This loading is low considering the decreased potency of paclitaxel compared to doxorubicin. Theoretically, paclitaxel or peptidyl derivatives will be released from the polymer by hydrolytic or enzymatic (esterase) degradation of the ester bond, or by proteolytic cleavage of the peptidyl linker. In the phase-I study, PNU166945 was administered by a 1-hour infusion every three weeks75.
HPMA copolymer-camptothecin (MAG-CPT; PNU 166148)
Camptothecins have poor solubility and their nonspecific toxicity has made them attractive candidates for polymer conjugation. The drug conjugation was an ester linkage. Camptothecin is released from this conjugate either by chemical or by esterase-mediated hydrolysis76.
HPMA copolymer-platinate (AP5280)
A library of HPMA copolymer-platinates synthesized as “cisplatin” or “carboplatin” mimetics have produced a clinical candidate. These conjugates were prepared from HPMA copolymer precursors containing either -Gly-Gly-ONp or Gly-Phe-Leu-Gly-ONp side chains (5 or 10 mol %). The side chains were modified by hydrolysis (-COOH) or aminolysis with ethylenediamine , aminomalonate, or aminoaspartate to provide the terminal ligands for platination. An HPMA copolymer Gly-Phe-Leu-Gly-en-Pt required lysosomal activation to release active platinum species, and this was confirmed by the observation that conjugates containing the nondegradable linker -Gly-Gly-en-Pt were completely inactive in vivo. Whereas the -COO-Pt released platinum species much too rapidly for useful delivery (i.e., it would simply fall apart in plasma after injection), the malonate derivative showed a slower, more useful rate of hydrolysis. Access Pharmaceuticals selected this compound for preclinical development77.
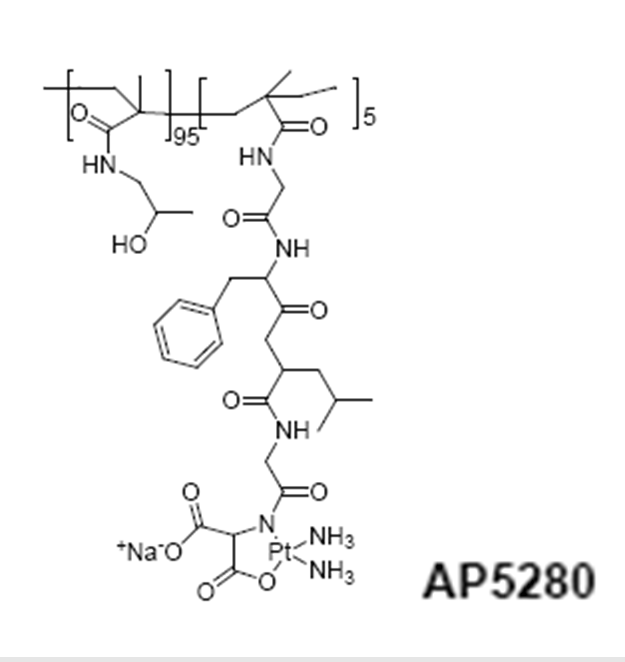
HPMA copolymer conjugation to adriamycin considerably decreased its toxicity against hematopeietic precursors in bone marrow in vitro and decreased its cardiotoxicity in rats78.
Poly(L-glutamic acid)—paclitaxel (CT-2103, XYOTAX)
The use of poly(L-glutamic acid) (PG) as an anticancer carrier was first proposed in the 1980s, but the PG-doxorubicin conjugate synthesized at that time did not progress to clinic. Use of this carrier was successfully revisited in the 1990s by Wallace et al.79, and a PG paclitaxel conjugate (acquired by Cell Therapeutics, Inc.), CT-2103, has been advancing successfully and very rapidly through an early clinical development program. In CT-2103, the paclitaxel is linked through the 2’ position to the ester bond to the carboxylic acid of PG. The conjugate has a molecular weight of approximately 40,000 and contains 37 wt% paclitaxel. Moreover, it is 80,000 more soluble than paclitaxel. This conjugate has the advantage of a biodegradable PG polymer backbone, and it has been found that exposure to cathepsin B results in liberation of diglutamyl-paclitaxel. Remarkable antitumor activity was seen in a variety of in vivo tumor models that, together with evidence of tumor targeting by the EPR effect, paved the way for clinical testing80. Interestingly, it has been shown in preclinical studies combining conjugate administration with radiation treatment that tumor targeting of PGpaclitaxel by the EPR effect is significantly increased, leading to enhanced antitumor activity. This has important implications for possible clinical development of this and other polymer-drug conjugates.
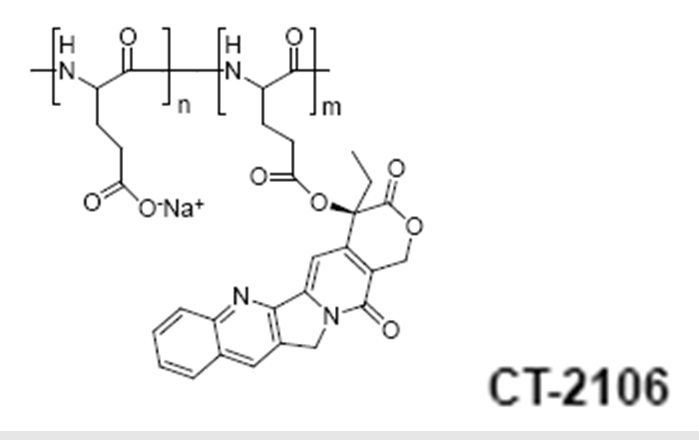
Poly(L-glutamic acid)-camptothecin (CT-2106)
Cell Therapeutics Inc. is also developing a camptothecin conjugate using the same PG polymeric carrier. Conjugates containing different linkers including -Gly, -Glycolic acid, Δ-Glu and b-Ala have been described81.
Polyethylene glycol-paclitaxel
In May 2001, Enzon82 reported the start of a phase-I clinical trial using a PEG-paclitaxel conjugates. The protocol has been designed to determine the safety, tolerability, and pharmacology of PEG-paclitaxel in patients with advanced solid tumors and lymphomas.
Dextran-doxorubicin (AD-70, DOX-OXD)
Polysaccharides have long been a popular choice for synthesis of polymer-drug conjugates2. Dextran (mainly Δ-1,6 polyglucose, with some Δ-1,4 branching) has been particularly popular owing to its clinical approval for use as a plasma expander. A dextran-doxorubicin conjugate (AD-70) was tested clinically. A dextran polymer with a molecular weight of approximately 70,000 was used to prepare the conjugate. Drug conjugation seemed to be by Schiff base formation using oxidized dextran also modified with glycine, as a pendant group for reaction with the anthracycline. The rationale of this conjugation approach was to utilize hypoxic conditions in the tumor to promote drug liberation83.
IT-101 is a camptothecin–polymer conjugate prepared by linking camptothecin (CPT) to a hydrophilic, cyclodextrin-based, linear polymer through ester bonds. These polymer conjugates with high molecular weights (90 000) have shown significant antitumor effects against human colon carcinoma xenografts.
The studies indicate that intravenous administration of IT-101, a cyclodextrin based polymer–CPT conjugate, gives prolonged plasma half-life and enhanced distribution to tumor tissue when compared to CPT alone. The data also show that active CPT is released from the conjugate within the tumor for an extended period. These effects likely play a significant role in the enhanced antitumor activity of IT-101 when compared to CPT alone84.
In the view of these findings, we planed to design and synthesize new polymeric derivative as possible Prodrugs for some well-known anticancer agents (e.g. Chlorambucil, guanazole and cytarabine).
References
1 B. Vogelstein and K. Kinzler, Cancer genes and the pathways they control. Nature Medicine, 10: 789-799 (2004).
2 J. D. Maltzman and M. D. Abramson; Cancer center of the University of Pennsylvania, posting date: November 5, (2003).
3 Zubrod, C. C;In Fundamentals of cancer chemotherapy; Hellman K, Carter S. K. (Eds); Mc Millan Publishing Company, New York, (1987); pp. 101-110.
4 Roads, C.P; Nitrogen mustard in the treatment of neoplastic disease; official statement, JAMA.
5 Callery, P. and Peter, G.: Cancer and cancer chemotherapy. In: Foye’s principles of medical chemistry (5th Ed.). David, A.W.and Thomas, L.L. (EDs), Lippincott, Williams & Wilkins, Philadelphia, (2002); p.924.
6 Price, C.C.: Chemistry of Alkylation's. In: Handbook of experimental pharmacology .Sartorelli, A.G. and Johns, D.J. (EDs), Springer Verlag, New York 1975; 38: part 2; p 4.
7 Connors, T.A: Mechanism of action of 2-chloroethylamine derivative, sulfur mustard, epoxides and aziridines. In: Handbook of experimental pharmacology. Sartorelli, A.G. and Johns, D.J. (EDs), Springer Verlag, New York 1975; 38: part 2; p 19.
8 Silverman, R.B.: The organic chemistry, dmg design and drug action. Academic Press, New York, 1992; p. 149
9 Montagomry, J.A.; et al: Drug for neoplastic disease. In Medicinal chemistry (3ed Ed). Burger, A. (ED), Wiley Interscience, New York, 1970; pp. 680.
10 Waskman, S.A. and Woodruff, H.B.: The actinomycines and their importance in the treatment of tumors in animals and man. Ann. N Y. Acad.Sci. 1960; 89: 287.
11 Rao, K.V.: A new antibiotic with anti-tumor activity. Antibiotic Chemotherapy 1962; 12: 182.
12 Mc Govern, J.P.: Pharmacological principle. In: Cancer chemotherapy (2nd Ed). Dorr, R.T. and Von Hof, D.D. (EDs), Norwalk,CT,Appeltone and Lange, 1994; pp. 15.
13 DiMarco, A.: Anthracycline antibiotics. In: Cancer Medicine (2nd Ed.) Lea and Febiger, Philadephia, 1982; p. 872
14 Crooke, S.T. and Brander, W.T.:Mitomycin C. ; a review . Cancer Treat. Rev. 1976, 3:121.
15 Stark, G.R. and Bartlett, P.A.: Design and use of potent and specific enzyme inhibitors. Pharmacol.Ther. 1983; 23:45.
16 Creasey, W.A.: Plant alkaloids. In: 5: A comprehensive treatise. Plenum Press, New York, 1977; p. 379.
17 Kiang, D.T. and Kennedy, B.J.: Tamoxifen (antiestrogen) therapy in advanced breast cancer. Honn.Res. 1962; 18: 387
18 Neri, R.; et al.: Endocrinology 1972; 18: 687.
19 Lote, K.; England, T.; Harber, B.; and Stenwing, B.: Survival, prognostic factors and therapeutic efficacy in low-grade glioma. J Neurooncol. (1997); 15: 275.
20 Chamberlain, C.L. and Karmanike, P.A.: Preclinical guidelines for the treatment of malignant glioma. West J. Med. (1998); 168: 1 14.
21 Eckhardt, S. CME J Gyn. One. 2001, 2, 312.
22 Dunagin WG: Clinical toxicity of chemotherapeutic agents. Semin Oncol 9: 14-22, 1982.
23 National Institutes of Health website. Available at: //www.nih.gov/.
24 Stella VJ, Himmelstein KJ. Prodrugs and site-specific drug delivery. J Med Chem. 1980; 23:1275-1282.
25 Stella V. Pro-drugs: an overview and definition. In: Higuchi T, Stella V, eds. Prodrugs as Novel Drug Delivery Systems. ACS Symposium Series. Washington, DC: American Chemical Society; 1975:1-115.
26 M.C. Garnett, Targeted drug conjugates: principles and progress. Adv. Drug Deliv. Rev. 53 (2001), pp. 171-216.
27 P. Rejmanova, J. Kope ek, J. Pohl, M. Baudy and V. Kostka, Polymers containing enzymatically degradable bonds. Chem. 184 (1983), pp. 2009-2020.
28 Brown; John M. (Redwood City, CA); Liu; Shie-Chau (Redwood City, CA); Giaccia; Amato J. (Stanford, CA); Minton; Nigel P. (Salisbury, GB): Anaerobe targeted enzyme-mediated prodrug therapy: January 10, 2006, United States Patent: 6,984,513.
29 Melton, R.G., et al., "Covalent Linkage of Carboxypeptidase G2 to Soluble Dextrans-II--In Vivo Distribution and Fate of Conjugates," Biochemical Pharmacology, (1987).36(1):113-121.
30 Kenneth D Bagshawe: Expert Review of Anticancer Therapy; 2006, Vol. 6, No. 10, pp 1421-1431.: VITOLS K. S. ; HAAG-ZEINO B. ; BAER T. ; MONTEJANO Y. D. ; HUENNENKENS: Methotrexate a.-phenylalanine : optimization of methotrexate prodrug for activation by carboxypeptidase A-monoclonal antibody conjugate; 1995,Cancer research , vol. 55, pp. 478-481.
31 Springer CJ, Antoniw P, Bagshawe KD, Searle F, Bisset GM, Jarman M. Novel prodrugs which are activated to cytotoxic alkylating agents by carboxypeptidase G2. J Med Chem 1990; 33:677-681.
32 Abramowitz, N., Schechter, I. & Berger, A. (1967) Bio- chem. Biophys. Res. Commun. 29, 862.
33 Baldino CM. Perspective articles on the utility and application of solution-phase combinatorial chemistry. J Comb Chem. 2000; 2:89- 103.
34 Brocchin i S, James K, Tangpasuthadol V, Kohn J, A combinatorial approach for polymer design. J Am Chem Soc. 1997:119:4553-4554.
35 Brocchin i S, James K, Tangpasuthadol V, Kohn J, A combinatorial approach for polymer design. J Am Chem Soc. 1997:119:4553-4554.
36 Sinha VR, Khosia L. Bioabsorbable polymers for impfantable therapeutic systems. Drug Dev Ind Pharm. 1998:24:1129-1138.
37 Ravi Kumar MN. Nano and microparticles as controlled drug delivery devices. J Pharm Pharm Sci. (2000): 3:234-258.
38 Langer R. Drug delivery and targeting. Nature. 1998,392:5-10.
39 Kopecek J، Kopeckova P, Minko T, Lu ZR. HPMA copolymer anticancer drug conjugates: Design, activity, and mechanism of action. Eur J Pharm Biopharm. (2000); 50:61-81.
40 Shiah JG, Kohak C, Spikes JD, Kopecek J . Solution and photoproperties of N-(2-hydroxypropyl) methacrylamide copolymer mesochlorin ee conjugates. J Phys Chem B. (1997): 101:6803-6809
41 Shiah JG, Kohak C, Spikes JD, Kopecek J. Influence of pH on aggregation and photoproperties of N-(2-hydroxypropyl) methacrylamide copolymer- mesochlorin QQ conjugates. Drug Delive1y. (1998):5:119-126.
42 Veronese FM, Morpurgo M. Bioconjugation m pharmaceutical chemistry. Farmaco. (1999):54:497-516.
43 Duncan R, Dimitrijevic S, Evagorou EG. The role of polymer conjugates in the diagnosis and treatment of cancer. STP Phaima Sci. (1996):6:237-263.
44 de Duve C, de Barsy T, Poole B, Trouet A, Tulkens P, Van Hoof F.Lysosomotropic agents. Biochem Pharmacol. 1974:23:2495-2531.
45 Petrak K, Goddard P. Transport of macromolecules across the capillary walls. Adv Drug Del Rev. 1989:3:191-214.
46 Ehrlich P. Studies in immunity. New York: Plenum Press; 1906.
47 Ringsdorf H. Structure and propetties of pharmacologically active polymers, J Polym Sci Polym Sup. 1975:51:135-153.
48 Williams AS, Camilleri JP, Goodfellow RM, Williams BD. A single intra-articular injection of liposomally conjugated methotrexate suppresses joint inflammation in rat antigen-induced arthritis. Br J Rheumatol. (1996), 35:719-724.
29 TorchiNnVP. Drug targeting. Eur J Pharm Sci. 2000; 11 Suppi 2:S81-91.
50 Torchilin VP, Zhou F, Huang L. pH-sensitive liposomes. Journal of Liposome Research. (1993); 3:201-255.
51 Orekhova NM, Akchurin RS, Belyaev AA, Smirnov MD, Ragimov SE, Orekhov AN. Local prevention of thrombosi s in animal arteries by means of magnetic targeting of aspirin-loaded red cells. Thromb Res. 1990:57:611-616.
52 Torchilin VP, Papisov Ml, Orekhova NM, Belyaev AA, Petrov AD, Ragimov SE. Magnetically driven thrombolytic preparation containing immobilized streptokinase-targeted transport and action. Haemostasis. (1988): 18:113-116.
53 Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. (2001):41:189-207.
54 Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Controlled Release. (2001):74:47-61.
55 Skinner SA, Tutton PJ, O'Brien PE. Microvascular architecture of experimental colon tumors in the rat. Cancer Res. 1990:50:2411-2417.
56 Suzuki M, Takahashi T, Sato T. Medial regression and its functional significance in tumor-supplyi ng host arteries. A morphometric study of hepatic arteries in human livers with hepatocellular carcinoma. Cancer. 1987:59:444 -450.
57 Steyger PS, Baban DF, Brereton M, Ulbrich K, Seymour LW .Intratumourai distribution as a determinant of tumour responsiveness to therapy using polymer-based macromolecular prod rngs. J Controlled Release. (1996); 39:35-46.
58 Folkman J. Tumor angiogenesis: Therapeutic implications. N Engl J. Med. 1971:285:1182-1186.
59 Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992:267: 10931- 10934.
60 Noguchi Y, Wu J, Duncan R, et al. Early phase tumor accumulation of macromolecules: A great difference in clearance rate between tumor and normal tissues. Jpn J Cancer Res. (1998):89:307-314.
61 Rihova B, Kopecek J. Biological properties of targetable poly[N-(2- hydroxypropyl)methacrylam ide] - antibody conjugates. J Controlled Release. 1985:2:289-310.
62 Flanagan P.A, Duncan R, Rihova B, Subr V, Kopecek; J. Immunogenicityof protein- N-(2-hydroxypropyl) methacrylamide copolymer conjugates measured in A/J and b lO mice. J Bioact Comp Polym. 1990:5:151-166.
63 H. Soyez, E. Schacht, S. Vanderkerken, The crucial role of spacer groups in macromolecular prodrug design, Adv. Drug Deliv. Rev. 21 (1996) 81- 106.
64 F. Kratz, U. Beyer, M.T. Schutte, Drug- polymer conjugates containing acid-cleavable bonds, Crit. Rev. Ther. Drug Carrier Syst. 16 (1999) 245- 258.
65 T. Roth, F. Kratz, C. Steidle, C. Unger, H.H. Fiebig, In vitro and in vivo antitumor efficacy of acid sensitive albumin and poly(ethylene) glycol conjugates of doxorubicin, Ann. Oncol. 9 (1998) 102.
66 F. Kratz, U. Beyer, M.T. Schutte, Drug- polymer conjugates containing acid-cleavable bonds, Crit. Rev. Ther. Drug Carrier Syst. 16 (1999) 245-288.
67 T. Etrych, P. Chytil, M. Jeh 'nkova', B. R hova', K. Ulbrich, Synthesis of HPMA copolymers containing doxorubicin bound via a hydrazone linkage. Effect of spacer on drug release and in vitro cytotoxicity, Macromol. Biosci. 2 (2002) 43- 52.
68 W.M. Choi, P. Kopecvkova ', T. Minko, J. Kopecvek, Synthesis of HPMA copolymer containing adriamycin bound via an acid-labile spacer and its activity toward human ovarian carcinoma cells, J. Bioact. Compat. Polym. 14 (1999) 447-456.
69 Kratz F, Beyer U, Schutte MT. Drug-polymer conjugates containing acid-sensitive bonds. Crit Rev Ther Drug Canier Syst. 1999; 16:245- 288.
70 W.C. Shen, H.J.P. Ryser, cis-Aconityl spacer between daunomycin and macromolecular carriers-a model of pHsensitive linkage releasing drug from a lysosomotropic conjugate, Biochem. Biophys. Res. Commun. 102 (1981) 1048-1054.
71 L.E. Gerweck, K. Seetharaman, Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer, Cancer Res. 56 (1996) 1194-1198.
72 Budman , H Calvert & E Rowinsky. Philadelphia, USA: Lippincott, Will iams & Wilkins. Polymer-drug conjugates. In Handbook of Anticancer Drug Development, pp 239-260.
73 Vasey P, Kaye SB, Monison R, et al (1999), Phase I clinical and pharrn acokinetic study of PK l [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents - drug-polymer conjugates. Clinical Cancer Research 5 83-94.
74 Duncan R. et al. (1989), J. Control Release 10: 51-63.
75 Meerum Terwogt J .M. et al. (2001), Phase I clinical and phannacokinetic study of PNU166945, Anticancer Drugs 12: 315- 323
76 Bissett D, Cassidy J, de Bono JS, et al: Phase Iand pharmacokinetic (PK) study of MAG-CPT (PNU 166148): a polymeric derivative of camptothecin (CPT). Br J Cancer (2004) 91:50-55.
77 Gianasi E. et al. (1999), Eur. J. Cancer 3: 994-1002.
78 Shiah JJ, Sun Y, Petreson CM, Kopecek J.: Biodestribution of free and N-(2-hydroxypropyl) methacrylamide copolymer-bound mesochlorin and adriamycin in nude mice bearing human ovarian carcinoma OVCAR-3 xenografts. J control release. (1999); 61:145- 157.
79 Li C. et al. (1998), Cancer Res. 58: 2404-2409.
80 Auzenne E. et al. (2002), Clin. Cancer Res. 8: 573-581.
81 Springett, G.M. et al.: Phase I study of CT-2106 (polyglutamate camptothecin) in patients with advanced malignancies. J. Clin. Oncol. (2004) 22, 3127.
82 Rowinsky EK, Rizzo J, Ochoa L, Takimoto CH, Forouzesh B, Schwartz G, Hammond LA, Patnaik A, Kwiatek J, Goetz A, Denis L, McGuire J, Tolcher AW: A phase I and pharmacokinetic study of pegylated camptothecin as a 1-hour infusion every 3 weeks in patients with advanced solid malignancies. J Clin Oncol (2003) 21:148-157.
83 Mitra, U. Gaur, P.C. Ghosh, A.N. Maitra, Tumour targeted delivery of encapsulated dextran - doxorubicin conjugate using chitosan nanoparticles as carrier, Journal of Controlled Release 74 (2001) 317-323.
84 Cheng J, Khin KT, Jensen GS, Liu A, and Davis ME:Synthesis of linear, beta-cyclodextrin-based polymers and their camptothecin conjugates. Bioconjug Chem (2003) 14:1007-1017.
© 2017 by the authors; licensee ARJ