ARJ 2016, 0(1), P.P. 13-22;
archives/2016/sabah_muthanna/
Research Article
Synthesis of New Ester Derivatives of Dexamethasone & Prednisolone as Possible Prodrugs
Muthanna S. Farhan* & Sabah J. Salih†
Abstract
New esters of Dexamethasone & Prednisolone were design, synthesized and evaluated as possible water-soluble prodrugs having higher partition coefficients with the aim of developing colon targeting of the drug and increasing the dose without increasing the solvent volume of injection. These esters are (2R,2'R)-((8S,9R,10R,11R,13S,14R,16S,17S)-9-fluoro-10,13,16-trimethyl-3-oxo-17-(2-((R)-5-oxotetrahydrofuran-2-carbonyloxy)acetyl)-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthrene-11,17-diyl) bis(5-oxotetrahydrofuran-2-carboxylate) (compound II) and (2R,2'R)-((8S,9S,10S,11R,13S,14R,16S,17S)-10,13,16-trimethyl-3-oxo-17-(2-((S)-5-oxotetrahydrofuran-2-carbonyloxy)acetyl)-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthrene-11,17-diyl) bis(5-oxotetrahydrofuran-2-carboxylate) (compound III).
Their lipophilicity (expressed as the log P value) of Dexamethasone & Prednisolone and their synthesized esters are investigated. These prodrugs revealed enhanced water solubility compared with Dexamethasone & Prednisolone base.
Compound (II) has higher partition coefficient than other synthesized esters, so expected to be higher partitioning through different route of administrations. Compound (III) appears to be the most promising synthesized prodrugs candidate as a parenteral delivery form of Prednisolone.
* Ph.D. Pharm. Chem, Department of Pharmaceutical Chemistry, College of Pharmacy, University of Baghdad.
† Ph.D. Pharm. Chem, Department of Pharmaceutical & Chemical analysis, NCDCR, Baghdad.
Key words: Dexamethasone, Prednisolone, prodrugs, tetrahydrofuran, thiazolidine & glutamic acid.
1. Introduction:
Steroid hormones & related products represents one of the most widely used classes of therapeutic agents. These drugs are used primarily in birth control, hormone-replacement therapy (HRT), inflammatory conditions and cancer treatment 1.
Dexamethasone & Prednisolone are synthetic corticosteroid drugs. Prednisolone is one of steroid hormones groups. Prednisolone, ∆1-hydrocortisone, 11β, 17, 21-trihydroxypregna- 1, 4-diene-3, 20-dione, has less salt-retention activity than hydrocortisone.
Because of low mineralocorticoid activity, it cannot be used alone for adrenal insufficiency. Prednisone is a prodrug that well converted by the liver into prednisolone.
Dexamethasone & Prednisolone acts as an anti-inflammatory and immunosuppressant. Dexamethasone potency is about 20-30 times that of Hydrocortisone and 4-5 times of Prednisolone.
This study reports the synthesis of Dexamethasone & Prednisolone esters as water-soluble prodrugs. The physicochemical properties and the lipophilicity (expressed as the log P value) of Dexamethasone & Prednisolone and its esters were investigated.

2. Experimental
All reagents and anhydrous solvents were of analar type and generally used as received from the commercial supplier (Merk_Germany, Sigma-Aldrich Germany &BDH_England). Dexamethasone & Prednisolone was supplied from National factory Lab. Chemicals and equipment - Jordan.
Melting points were determined by capillary method on Thomas Hoover apparatus (England) and ascending thin layer chromatography (TLC) was run on DC-Kartan SI Alumina 0.2 mm to check the purity and progress of reaction. The identification of compounds was done using iodine vapor and the chromatograms were eluted by:
A/ H2O: Methanol: Ether: Methylene chloride (1.5:8:15:77);
B/ Methylene chloride: Methanol (180:16)1.
IR spectra were recorded on model 500 scientific IR spectrophotometer, Buck Company (USA) as a KBr film.
CHN microanalysis has been done using Carlo Erba elemental analyzer 1106 was carried out at faculty of Pharmacy in University of Cairo.
2.1 Synthesis of 2-furancarboxylic acid, tetrahydro-5-oxo (comp. I):
A solution of sodium nitrate (12.6g, 0.183 moles) in distilled water (30ml) and sulfuric acid (73ml, 2N) were added drop wise simultaneously from separatory funnels to a suspension of L-glutamic acid (18g, 0.122 moles) in distilled water (50ml) under vigorous stirring. After the addition is complete, the clear solution was stirred at room temperature for an additional 15 hr and water is removed under reduced pressure with rotary evaporator.
The resulting pasty solid is triturated with a boiling acetone (75ml) and the hot solution is removed by decantation and set aside to cool. This operation is repeated four times. Removal of solvent with a rotary evaporated to give slightly yellow oil2. The viscous oil was crystallized twice from ethyl acetate-benzene-petroleum ether [60 - 80 °C] (1:1:1)3. The percent yield, physical data and Rf values of compound I are given in table (1).
2.2 Synthesis of : (2R,2'R)-((8S,9R,10R,11R,13S,14R,16S,17S)-9-fluoro-10,13,16-trimethyl-3-oxo-17-(2-((R)-5-oxotetrahydrofuran-2-carbonyloxy)acetyl)-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthrene-11,17-diyl) bis(5-oxotetrahydrofuran-2-carboxylate) (compound II).
Compound I (3.9g, 0.03 moles) was dissolved in dry tetrahydrofuran (30ml), and mixed with a solution of (8S,9R,10R,11R,13S,14R,16S,17S)-9-fluoro-11,17-dihydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17-dodecahydro-3H-cyclopenta[a]phenanthren-3-one (3.9g, 0.01 moles) in dry tetrahydrofuran (30ml). The mixture was cooled down to 0 C and a suspension of N, N-dicyclohexylcarbodiimide (DCC) (8.28g, 0.04 mole) in dry methylene chloride was added, with continues stirring at about 0 °C for 40 hours. N, N-dicyclohexyl urea (DCU) was filtered off. The filtrate was washed with ethyl acetate. The filtrate was concentrated under vacuum and the residue was redissolved in ethyl acetate, and the excess DCU which was still adhesive on the ester residue was precipitated out and filtered off. The clear filtrate was washed twice with 5% sodium carbonate solution, 0.1N HCl, and once with distilled water. The ethyl acetate layer was dried with anhydrous sodium sulfate and evaporated in vacuum. The resulted product was collected, recrystallized from methanol-ether to give compound II4. The percent yield, physical data and Rf values were given in table (1). CHO Calculated (C37H41FO14): C, 60.98; H, 5.67; O, 30.74. Found: C, 59.39; H, 5.55; O, 29.329.
2.3 Synthesis of (2R,2'R)-((8S,9S,10S,11R,13S,14R,16S,17S)-10,13,16-trimethyl-3-oxo-17-(2-((S)-5-oxotetrahydrofuran-2-carbonyloxy)acetyl)-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthrene-11,17-diyl) bis(5-oxotetrahydrofuran-2-carboxylate) (compound III).
Compound I (3.9g, 0.03 moles) was dissolved in dry tetrahydrofuran (30ml), and mixed with a solution of (8S,10S,11R,13S,14R,16S,17S)-11,17-dihydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-3-one (3.6g, 0.01 moles) in dry tetrahydrofuran (30ml). The mixture was cooled down to 0 C and a suspension of N, N-dicyclohexylcarbodiimide (DCC) (8.28g, 0.04 mole) in dry methylene chloride was added, with continues stirring at about 0 °C for 40 hours. N, N-dicyclohexyl urea (DCU) was filtered off. The filtrate was washed with ethyl acetate. The filtrate was concentrated under vacuum and the residue was redissolved in ethyl acetate, and the excess DCU which was still adhesive on the ester residue was precipitated out and filtered off. The clear filtrate was washed twice with 5% sodium carbonate solution, 0.1N HCl, and once with distilled water. The ethyl acetate layer was dried with anhydrous sodium sulfate and evaporated in vacuum. The resulted product was collected, recrystallized from methanol-ether to give compound III5. The percent yield, physical data and Rf values were given in table (1). CHO Calculated (C36H40O14): C, 62.06; H, 5.79; O, 32.15. Found: C, 60.136; H, 5.756; O, 31.346.
2.3 Standard Curve Preparation:
A series of different concentrations of chloroform solutions range from 0.01 mg/ml to 0.16 mg/ml for compounds II and III prepared from their corresponding stock solutions6.The absorbance of these solutions was obtained using ultraviolet spectrophotometer at λ240nm7.By plotting their absorbance versus corresponding concentrations, the standard curve was constructed for the compounds II and III. The values are recorded in table (2).
2.4 Partition Coefficient Estimation:
Partition coefficient of compounds II and III were estimated by taking 10ml of 0.1mg\ml chloroform solution of each of these compounds and mixed with an equal volume of distilled water in a separatory funnel, then shacked well for 30min, left for 10min in order to separate the two layers. Next step is collection and measuring the absorbance of chloroform layers at λ240nm8.By interpretation of the absorbance obtained by standard curve, we got the corresponding concentration of the compounds (amount remained in chloroform layer), and then by subtracting the concentration remained from the original concentration we got the concentration (amount) at the aqueous layer, as illustrated in table (3). The partition coefficient P values calculated by equation below9:
P = Concentration in Aqueous Layer/Concentration in Organic Layer …..Eq.1
The values are recorded in table (4).
3. Results and discussion:
The general routes outlined in schemes 1and 2 were used to synthesize all compounds described here. The purity was assessed by TLC and the assignments of the structures were based on elemental and spectroscopic methods of analyses. The IR spectral data revealed the characteristic carbonyl stretching vibration bands at 1725±1750 cm−1 of the ester of compounds II and III.
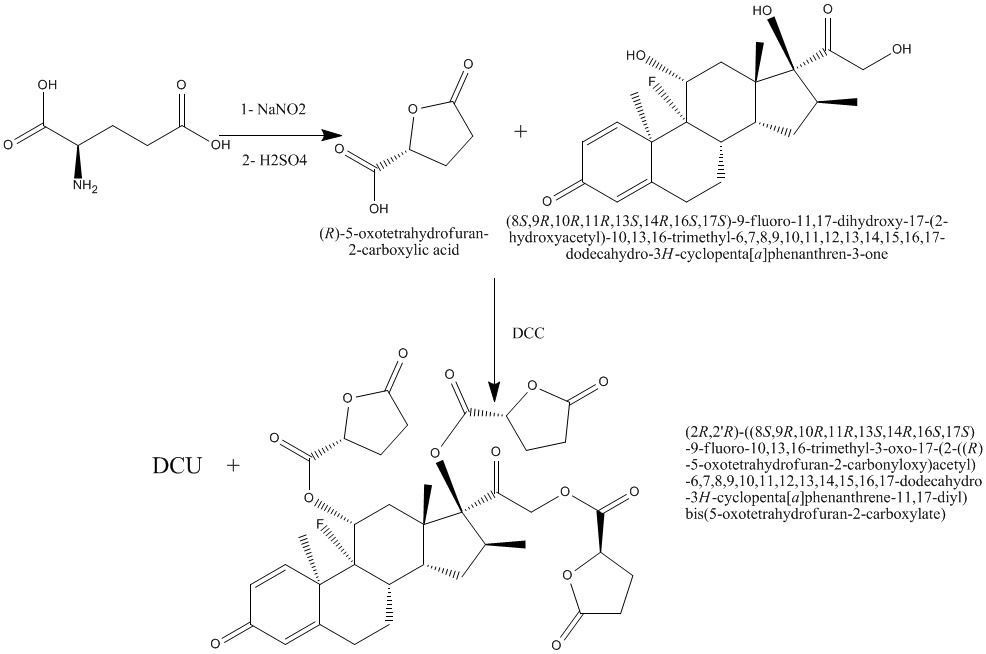
Scheme 1: Synthesis of compounds I & II
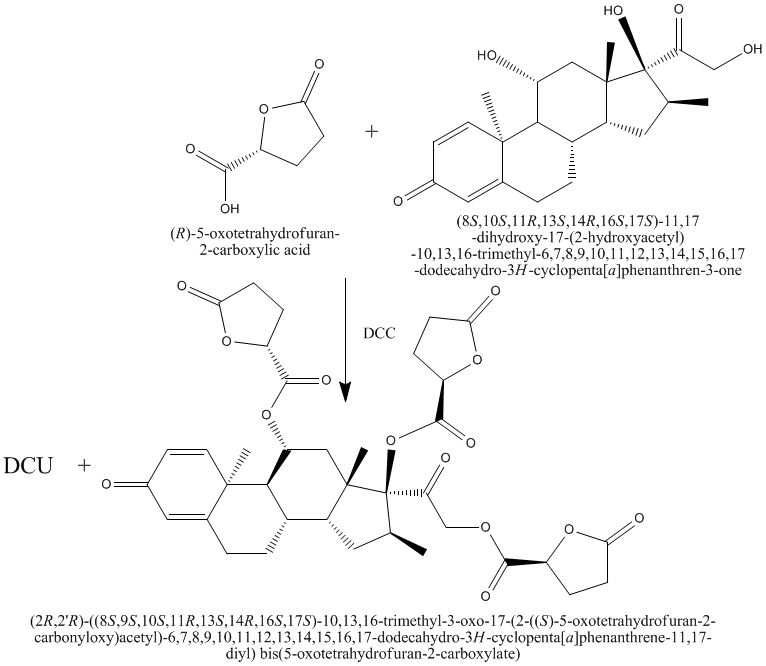
Scheme 2: Synthesis of compound III
Table (1): The characterization and physical data of the synthesized compounds.

Solvent systems are: A/ H2O: Methanol: Ether: Methylene chloride (1.5:8:15:77);
B/ Methylene chloride: Methanol (180:16).
Table (1B): The characterization and physical data of the synthesized origin compounds.
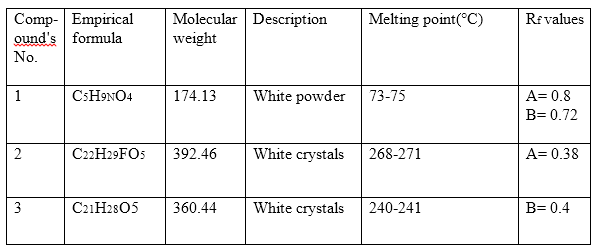
Solvent systems are: A/ H2O: Methanol: Ether: Methylene chloride (1.5:8:15:77);
B/ Methylene chloride: Methanol (180:16).
Partition coefficient determination:
After plotting the absorbance versus corresponding concentrations of the final compounds II & III, the standard curve was constructed for these compounds as illustrated in table (2)
Table 2: Absorbance of final compounds for each concentration.

Table 3: Absorbance and Concentration in organic layer & in aqueous layer of final compounds.
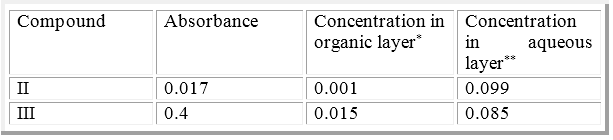
*(from standard curve),**(0.1- organic concentration)
Table 4: P & Log P values of initial and final compounds.

4. Conclusions:
The designed compounds have been synthesized successfully and their structures were confirmed. Compound II has higher partition coefficient than its base and compound III and so expected to be higher partitioning through different route of administrations. Compound III has more water solubility than its base so it expected to need less injectable formula.
References
1 Stahl, Chromatography, 1962, p 79-101.
2 Clayton, J. P, et al, Lactonyl Esters of Penicillins, J. Med. Chem, 1976, Vol.19, Issue 12, P.1385-1319.
3 Mori, K, Synthesis of Optically Active Forms of Sulcatol,Tetrahydron, 1975, Vol. 31, P.3011-3012.
4 Kovach, I. M, et al, Amino Acid Esters of Phenols as Prodrugs, J. Pharm. Sci, 1981, vol. 70, No. 8, p 881-885.
5 Kovach, I. M, et al, Amino Acid Esters of Phenols as Prodrugs, J. Pharm. Sci, 1981, vol. 70, No. 8, p 881-885.
6 Leo, A., Partition coefficients and Their Uses, Chem. Rev, 1971, Vol. 71, No. 6, P.525-616.
7 Maheshwari, R.K., et al., Novel Spectrophotometric Estimation, India. J. Pharmaceut. Sci., 2006, No. 2, P. 195-198.
8 Watson, D.G., Pharmaceutical Analysis, 2001, 2nd.Ed, Churchill
Livingstone Press, U.K, P.85.
9 Martin, A, Physical Pharmacy, 1993, 4th. Ed, Leo & Febiger, London,
U.K, P.241-242.
© 2016 by the authors; licensee ARJ